

Séquençage de l’ADN
CE GUIDE EST EN COURS DE CONSTRUCTION
Préfixe
Ce guide permet de commencer à explorer les vastes possibilités offertes avec l’accès au génome complet de la Caille Coturnix Japonica.
Lien pour les données génomique complètes

Pourquoi le séquençage est important?
Les oiseaux représentent la classe d’animaux domestiqués la plus répandue au monde et font l’objet de nombreuses études évolutives, biologiques et pathologiques qui illustrent les relations entre ces espèces aviaires.
Le moment de la maturité sexuelle est critique pour les plantes et les animaux.
Les cailles ont un programme de maturation unique par rapport aux autres oiseaux et atteignent la maturité sexuelle en très peu de temps.
Il as été détecté quatre gènes prometteurs pour ce trait sous sélection positive dans la voie de signalisation “GnRH” chez la caille.
Les gonadotrophines agissent sur les testicules et les ovaires pour favoriser leur développement et la production d’hormones stéroïdes.
Une analyse fonctionnelle plus poussée de ces gènes devrait fournir de nouvelles informations sur les mécanismes génétiques qui régulent la maturité sexuelle des oiseaux.
L’analyse des gènes et des mutations liés au développement et à l’évolution des caractères agronomiques chez la caille permettra également d’améliorer notre compréhension de la génétique de la domestication.
Des comparaisons à l’échelle du génome de lignées domestiquées (lignées de type pondeuse et de type viande) avec des cailles sauvages ont identifié plusieurs empreintes de sélection artificielle.
Ces régions de balayage sélectif abritent des gènes candidats associés à d’importants traits agro-économiques.
Les variations génétiques de ces gènes seront une ressource riche pour améliorer la production d’œufs et de viande de caille via la sélection génétique.
Il convient de noter que les cailles de type pondeuse et de type viande ne partageaient pas de régions de balayage sélectif par rapport aux cailles sauvages, ce qui signifie que les cailles de type pondeuse/de type viande pourraient avoir été sélectionnées indépendamment après la domestication ou qu’il y avait étaient deux événements de domestication distincts chez les cailles.
D’autres études sont nécessaires pour décrire complètement l’histoire de la domestication des cailles.
Sur la base des données de re-séquençage, il as été également identifié un haplotype totalement corrélé au contrôle de la couleur du plumage « marron/jaune », un trait qui a été largement utilisé dans l’élevage de cailles domestiques en tant que marqueur lié au sexe.
Certaines études récentes qui utilisent des données génomiques soutiennent notre compréhension actuelle de la phylogénie des familles Perdicinae, Meleagridinae et Phasianinae.
Cependant, sans données à l’échelle du génome, il n’est pas possible de tirer des conclusions solides.
Ici, des séquences du génome entier de caille, de dinde et de poulet japonais ont été utilisé pour représenter chaque clade et résoudre les relations phylogénétiques entre les familles Perdicinae, Meleagridinae et Phasianinae.
Cette étude a fourni des branches entièrement résolues avec des données à l’échelle du génome, soutenant une scission des branches Perdicinae et Phasianinae de la branche Meleagridinae d’environ 69 Millions d’Années.
Un étalonnage basé sur des fossiles de manchots primitifs, ainsi que sur des séquences du génome mitochondrial d’un albatros moderne (Diomedea melanophris), d’un pétrel (Pterodroma brevirostris) et d’un plongeon (Gavia stellata), a permis d’estimer le temps de divergence des Ansériformes et des Galliformes à 77,1± 2,5 Millions d’Années.
D’autres données récentes sur le génome aviaire ont été utilisées pour estimer la divergence des Ansériformes et des Galliformes à environ 66 Millions d’Années.
La résolution de leur phylogénie améliorera notre compréhension de la génétique de la spéciation de la caille, du poulet et de la dinde.
Dans cette recherche, les chercheurs ont obtenu une ébauche de haute qualité du génome de la caille japonaise et des données de re-séquençage du génome entier de plusieurs sous-populations de cailles, qui offriront de nouvelles opportunités pour mieux comprendre la biologie de la caille, et développer des marqueurs moléculaires pour améliorer les traits agronomiques économiquement importants.

Caractéristiques du génome de la caille
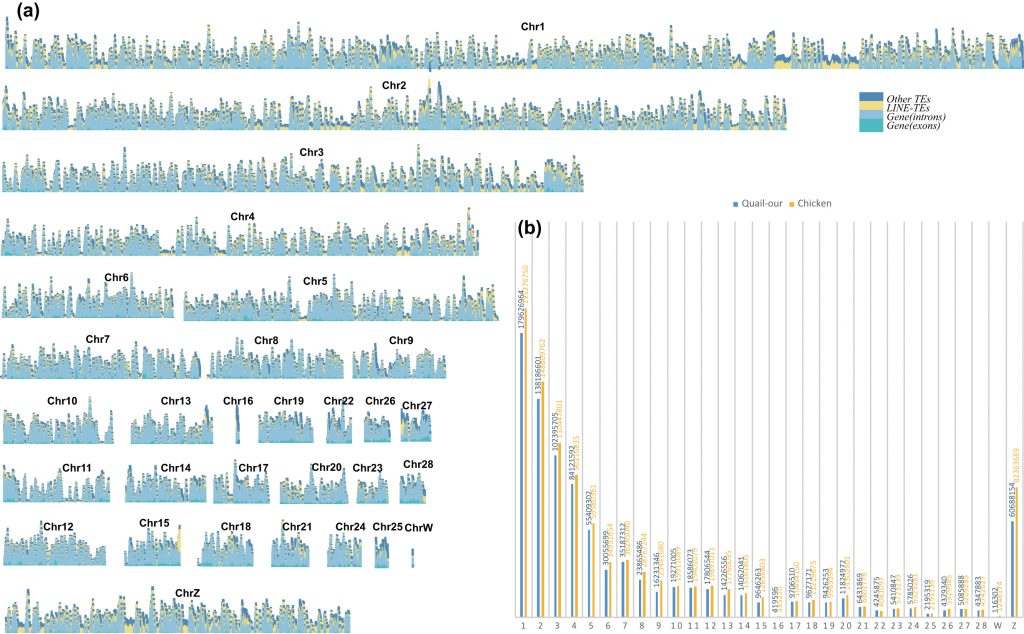
Un ADN génomique de haute qualité extrait d’une caille femelle (Shendan Quail 1) a été utilisé pour générer 262 Gb de séquence (couverture d’environ 238 fois de l’ensemble du génome) à l’aide de la plateforme Illumina HiSeq 2000.
Le génome assemblé à l’aide de SOAPdenovo2 s’étend sur 1,04 Gb (93,9 % de la taille estimée du génome de la caille) avec des longueurs contig N50 et d’échafaudage N50 de 27,9 kb et 1,8 Mb, respectivement.
Environ 901 Mo de séquence (86,6 % de l’ensemble du génome) ont été ancrés sur 30 chromosomes à l’aide d’une carte de liaison génétique précédemment rapportée.
Ces chercheurs ont aligné ces chromosomes sur un assemblage de génome de caille précédemment rapporté (NCBI BioSample : SAMN03989050) et ont constaté que les deux génomes avaient un degré élevé (92,14 %) de cohérence.
La longueur et la distribution GC des chromosomes sont également très cohérentes entre les séquences du génome de la caille et du poulet.
Pour évaluer la qualité du génome de caille assemblé, sept clones de fosmides (chacun d’environ 40 kb de long) ont été séquencés et cartographiés vers l’assemblage du génome de la caille avec un taux de couverture élevé (> 92 % pour tous, et six des sept fosmides > 98,4 %).
Pour évaluer l’intégrité des gènes codant pour les protéines dans l’assemblage du génome de la caille, tous les transcrits assemblés à partir des données RNA-Seq échantillonnées à partir de l’hypothalamus et de l’ovaire à trois stades de maturité de la caille (avant ponte [BL], ponte [L] et pic -pose [LP]) ont été mappés sur le génome assemblé, et ∼96,33% du total des gènes BUSCO complets peuvent être identifiés dans le génome.
Ces mesures ont démontré la haute qualité de ces assemblage génomique, lui permettant de servir de génome de référence pour les recherches ultérieures sur le génome de la caille.
Crédit: Population genomic data reveal genes related to important traits of quail

Relations évolutives au sein de la famille des Phasianidae
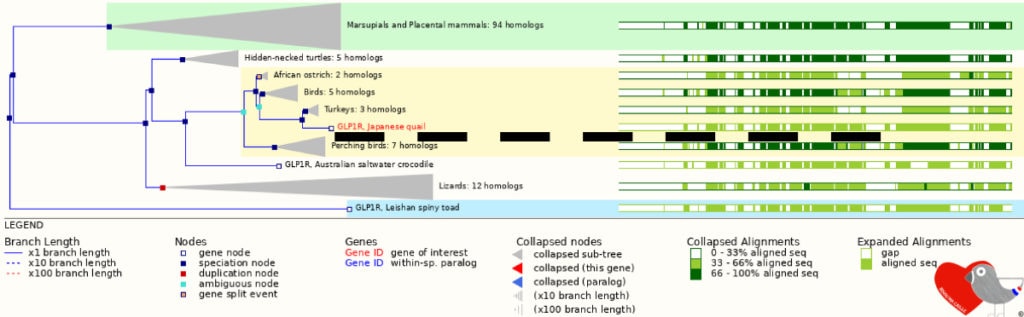
Pour résoudre le débat phylogénétique dans la famille des Phasianidae et établir la position phylogénétique de la caille par rapport aux autres espèces aviaires, ces chercheurs ont défini 12 178 familles de gènes chez la caille et 10 autres espèces d’oiseaux représentatives, avec Alligator sinensis (alligator chinois) servant d’exogroupe.
Au total, 9 631 familles de gènes ont été partagées entre quatre espèces (Taeniopygia guttata, Pseudopodoces humilis, Gallus gallus et Coturnix japonica), et 4 393 orthologues à copie unique ont été partagés entre 12 espèces.
Ces gènes orthologues à copie unique ont été utilisés pour construire un arbre phylogénétique et estimer les temps de divergence de la caille par rapport aux autres oiseaux.
La caille a été mappée à la branche évolutive contenant la volaille domestiquée et était la plus étroitement liée à la lignée de poulet, partageant un ancêtre commun d’environ 22,2 Millions d’Années .
Ces chercheurs ont utilisé des données comparatives à l’échelle du génome pour estimer le temps de divergence des Galliformes et des Ansériformes à 69,1 (64,5–75,4) Millions d’Années.
Leurs résultats soutiennent donc pleinement une relation plus étroite entre la caille et le poulet qu’entre la caille et la dinde.
La phylogénie que ces chercheurs ont générée impliquait que les génomes de la caille et du poulet partagent probablement des similitudes significatives, ce qui rend la comparaison de leurs génomes intrigante.
Crédit: Population genomic data reveal genes related to important traits of quail
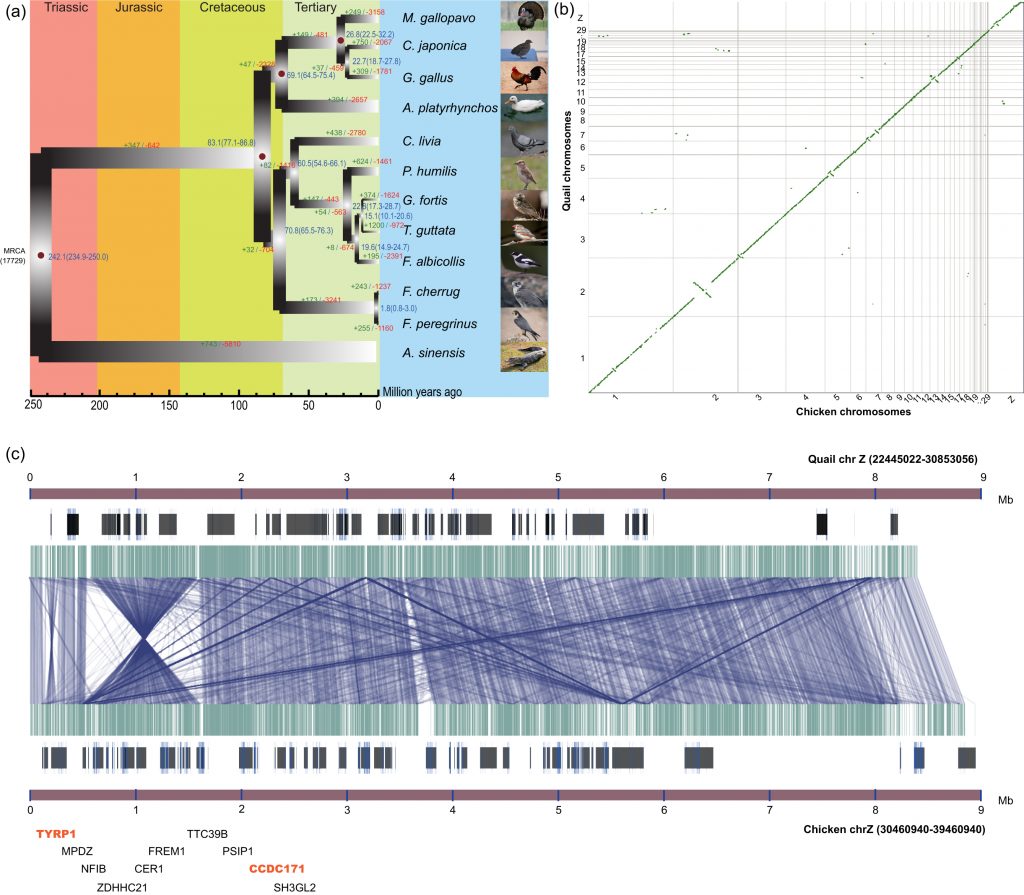
Au total, 95,5% des séquences du génome de la caille se sont produites dans des blocs colinéaires avec ceux du poulet.
Cependant, un total de 131 grandes inversions (longueur de bloc > 5 kb) entre les chromosomes de la caille et du poulet ont également été identifiées et la plupart d’entre elles étaient localisées sur les chromosomes 1 (24 points de rupture) et Z (24 points de rupture).
Ensuite, pour étudier la nature des cassures chromosomiques qui différencient les génomes de la caille et du poulet et pour associer ces différences à d’éventuels changements phénotypiques au cours de leur divergence, ces chercheurs ont testé des enrichissements génétiques aux frontières.
Ces chercheurs ont identifié 433 gènes situés dans les régions de 1 kb flanquant les points de rupture de ces inversions.
Ils ont testé l’enrichissement de la fonction génique au sein de ces inversions et recherché des mutations candidates qui pourraient contribuer à des phénotypes spécifiques chez la caille par rapport au poulet.
Les résultats de l’analyse d’enrichissement des termes GO de ces gènes ont révélé que les termes GO:0005882 : filament intermédiaire (P = 1,53e-05) et GO : 0005200 : constituant structurel du cytosquelette (P = 0,00029) étaient significativement enrichis.
En particulier, un gène codant pour la protéine 1 liée à la tyrosinase (TYRP1) a été identifié dans la région flanquante d’une inversion sur le chromosome Z, qui a été signalé comme un locus candidat pour le phénotype récessif du roux (br(r)) lié au sexe chez la caille du Japon.
Gènes impliqués dans la maturité sexuelle précoce
Pour explorer le mécanisme biologique de la maturité sexuelle très précoce chez les cailles, des gènes ont été tracés à partir à la fois de l’évolution de la famille de gènes et des événements de sélection positive dans la lignée des cailles.
Ces chercheurs ont constaté que plusieurs familles de gènes se sont développées dans le génome de la caille par rapport à celui d’autres oiseaux domestiqués.
Ces familles comprennent celles codant pour la gonadolibérine 1 (GnRH1), le domaine catalytique de la lysophospholipase et la phospholipase A2.
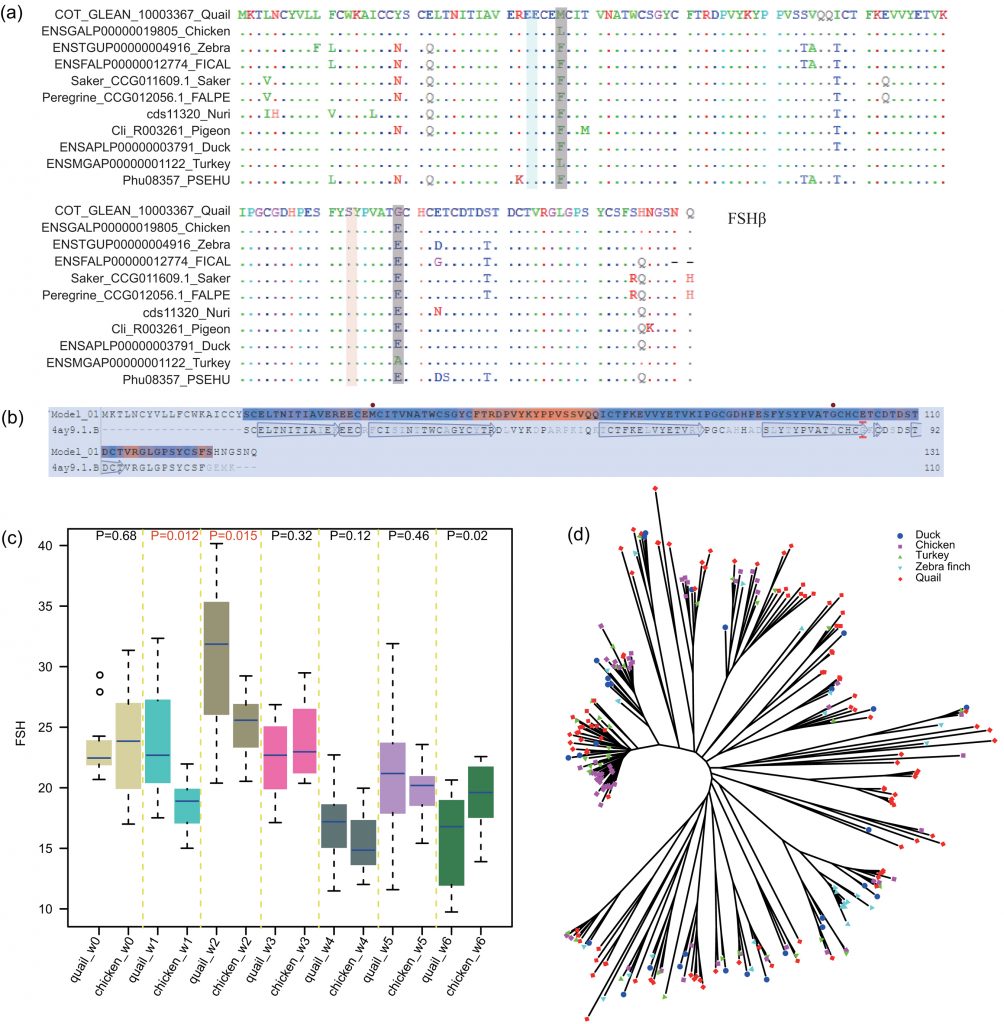
De plus, quatre gènes sélectionnés positivement (PSG) ont été détectés dans la lignée des cailles, et les protéines codées par ces gènes (FSHβ, PLCB4, ITPR1 et PLA2G4) sont impliquées dans la voie de signalisation de la GnRH.
La protéine FSHβ est une hormone polypeptidique glycoprotéique qui, associée à l’hormone lutéinisante, contribue à la croissance et à la reproduction.
La transcription du gène FSHβ est l’étape limitante de la synthèse hormonale requise pour la folliculogenèse ovarienne chez les femelles et pour la spermatogenèse chez les mâles, en association avec la testostérone.
Ils ont identifié deux acides aminés dans la protéine FSHβ de caille en position 37 (M→F/L) et en position 99 (G→E/A) qui étaient censés être sous sélection positive.
Ils ont utilisé ELISA pour mesurer le niveau de protéine FSHβ au cours des premiers stades de développement et ont constaté que le niveau de FSHβ dans le sang de cailles à maturation précoce est systématiquement plus élevé que celui du poulet (P < 0,05).
Ces chercheurs ont utilisé le SWISS-MODEL pour modéliser la structure de la FSHβ de caille en utilisant la protéine Follitropin subunit beta (4ay9.1.B) comme modèle.
Ces deux substitutions d’acides aminés ont été cartographiées sur la structure protéique 3D et étaient situées près de la feuille plissée qui interagit avec le récepteur de la FSH.
PLCB4, ITPR1 et PLA2G4, ainsi que d’autres molécules (par exemple, l’inositol 1,4,5-trisphosphate, le diacylglycérol et la protéine kinase C) stimulent la libération de gonadotrophines, notamment l’hormone lutéinisante et la FSH.
Les expansions de gènes dans les familles GnRH, ainsi que les PSG dans la voie de signalisation GnRH, sont susceptibles d’être impliquées dans l’accélération de la croissance et de la maturité sexuelle chez la caille.
Par la suite, ils ont scanné à la fois les SNP synonymes et non synonymes trouvés dans la séquence codante (CDS) de ces quatre gènes chez les 31 individus sauvages et domestiques et avons constaté que tous les 83 SNP sauf quatre étaient des substitutions synonymes.
Cependant, tous les allèles divergents des loci SNP ne se sont généralement pas séparés selon les trois sous-populations, et les cailles domestiques et sauvages n’ont présenté aucun balayage sélectif à grande échelle autour de ces gènes.
Crédit: Population genomic data reveal genes related to important traits of quail
Familles de gènes liées à la fonction du système immunitaire
Les chercheurs ont identifié un total de 1 587 gènes liés à la réponse immunitaire chez la caille en alignant l’ensemble des gènes prédits de la caille contre 2 257 gènes qui ont été annotés avec des rôles dans les réponses immunitaires innées d’Homo sapiens, Mus musculus ou Bos taurus. dans les bases de données InnateDB ou GO.
Par rapport au poulet, à la dinde et au canard, plusieurs familles de gènes étendus ont été identifiées chez la caille.
Ceux-ci comprenaient Klf4, qui est indispensable pour la différenciation des monocytes inflammatoires et la régulation négative de la réponse immunitaire innée contre plusieurs virus dans les cellules 293 du rein embryonnaire humain (22 copies chez la caille, 17 chez le poulet, 16 chez la dinde et 12 chez le canard); Foxa2, qui régule les programmes génétiques qui influencent l’inflammation pulmonaire médiée par les cellules Th2 (13 copies chez la caille, 7 chez le poulet, 4 chez la dinde et 5 chez le canard) ; et ITCH, qui agit dans la différenciation des cellules T auxiliaires et l’activation et la tolérance des cellules T (7 copies chez la caille, 3 chez le poulet, 4 chez la dinde et 2 chez le canard).
De plus, ils se sont concentrés sur le nombre de gènes du «sous-type d’immunoglobuline» et ont constaté qu’il y en avait 109 chez la caille, tandis que le poulet, le canard, le mandarin diamant (Taeniopygia guttata) et la dinde avaient chacun 62 ou moins de gènes liés aux immunoglobulines (62 copies chez le poulet , 29 chez le canard, 29 chez le diamant mandarin et 34 chez la dinde).
Ces chercheurs ont détecté 69 gènes codant pour un « domaine de transcriptase inverse ou de transcriptase inverse » putatif chez la caille, mais n’ont trouvé que deux et quatre de ces gènes chez le poulet et la dinde, respectivement.
Ces domaines sont des signatures de rétrovirus intégrés dans le génome de l’hôte.
Ensuite, ils ont comparé la région du CMH-B entre la caille et le poulet et avons constaté qu’il y avait une inversion incluant les gènes codant pour les protéines TAP1 et TAP2, qui transportent les peptides du cytosol dans le réticulum endoplasmique pour lier les molécules du CMH de classe I. en cours de synthèse, et BFIV21, qui code pour une protéine qui présente des antigènes comme le virus de la leucose aviaire.
Ils ont également trouvé quatre copies du gène BLEC2 (récepteur des cellules NK de type lectine de type C) chez la caille, mais une seule chez le poulet.
Cependant, plusieurs autres gènes du CMH (par exemple, KIFC1, V-BG1 et BG2) n’ont pas été détectés dans le génome de la caille.
Une meilleure compréhension de ces changements génétiques liés au système immunitaire pourrait aider à caractériser la réponse immunitaire chez les cailles et faciliter le développement de vaccins ciblés pour les cailles.
Familles de gènes liées à la fonction comportemental “Tireur Arrière”
Le mutant comportemental “Tireur Arrière” de la caille japonaise se caractérise par une action de s’accroupir et de tirer vers l’arrière avec le cou se pliant ventralement et occasionnellement roulant vers l’avant. Le comportement anormal apparaît de l’éclosion à l’âge de 8 semaines. Les “Tireurs Arrière” ont été classés en trois types : (1) ceux présentant l’anomalie à l’éclosion et meurent dans les 5 jours ; (2) ceux qui présentent l’anomalie après 2 semaines d’âge et décèdent peu de temps après ; et (3) les survivants de longue date souffrant d’un comportement anormal et montrant une récupération progressive. Les mâles “Tireur Arrière” sont infertiles lorsqu’ils présentent l’anomalie, mais peuvent produire une descendance après la guérison. En revanche, les femelles ont tendance à avoir une durée de vie courte et manquent de capacité de reproduction. Des analyses génétiques ont indiqué que ce caractère mutant est contrôlé par 2 paires de gènes autosomiques récessifs.
Analyse d’association à l’échelle du génome de la couleur du plumage
Pour identifier les gènes liés au sexe conférant la couleur du plumage, les chercheurs ont élevé un ensemble de cailles de type pondeuse avec un plumage type Sauvage (marron) ou Isabelle (jaune), un trait qui a été confirmé comme ayant un héritage lié au sexe chez la caille et qui correspond à un rapport de ségrégation mendélienne, selon leur enquête précédente.
À partir de cet ensemble, ils ont échantillonné 40 cailles, dont 20 mâles et 20 femelles, et re-séquencé leurs génomes pour une analyse cas-témoins.
Ils ont identifié ∼20 Mbi-alléliques SNPs dans ces 40 cailles à une profondeur de séquençage de 20 à 30×.
Après contrôle de la qualité des SNP et de la LD redondante, un total final de 864 292 SNP a été retenu pour les analyses ultérieures.
Un test de similitude du génome des 40 échantillons de cailles a été mené à l’aide de SNP de haute qualité, et ils ont constaté que les similitudes entre n’importe quelle paire d’individus variaient de 70,4 % à 86,5 %, indiquant une homologie relativement élevée entre eux.
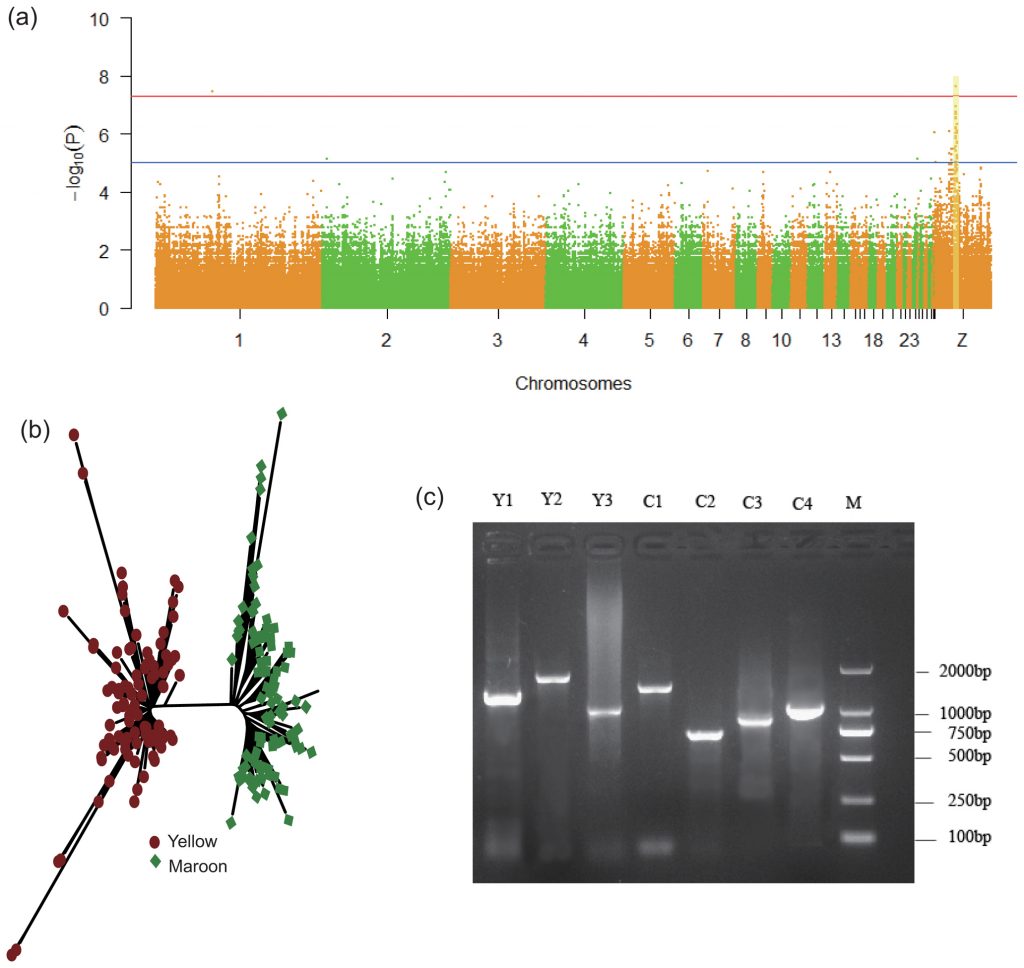
Crédit: Population genomic data reveal genes related to important traits of quail
En raison des relations relativement étroites entre les 40 échantillons, l’effet de la matrice de relativité affectant la variance de la couleur du plumage a été considéré comme la covariance.
Ces chercheurs ont évalué la matrice de relations des 40 échantillons en utilisant GEMMA v0.94 [67] et adopté le modèle linéaire mixte pour l’analyse d’association.
Par correction de Bonferroni, l’analyse d’association a montré que deux SNP, 61102026 sur le chromosome 1 et 23173971 sur le chromosome Z, avaient des effets significatifs sur la couleur du plumage (P ajusté = 0,028 et P = 0,019, respectivement).
Cependant, contrairement au locus sur le chromosome 1, les SNP sur le chromosome Z près de 23173971 ont montré un pic continu sur le graphique de Manhattan.
Dans leur analyse précédente de l’hérédité de la couleur du plumage, ils ont suggéré que le locus sur le chromosome Z était très probablement associé à la couleur du plumage.
Dans une étude de confirmation, ils ont ajouté les 21 cailles précédemment reséquencées au plumage « marron » (dont 10 cailles sauvages et 11 cailles de type pondeuse) pour réexécuter l’analyse d’association pour les deux loci.
Le locus sur le chromosome Z s’est avéré plus significativement associé à la couleur du plumage qu’auparavant (P ajusté = 0,015).
Inversement, il n’y avait pas de signal significatif sur le chromosome 1 (la valeur P ajustée pour le SNP 61102026 est tombée à 1 000).
Par la suite, ils ont trouvé que le SNP 23173971 sur le chromosome Z était proche du gène contenant le domaine Coiled-coil 171 (CCDC171) avec une longueur de 135 kb.
Par conséquent, ils ont choisi le SNP 23173971 sur le chromosome Z comme SNP index dans la région de 200 kb pour les tests d’association conditionnels basés sur les haplotypes.
Par conséquent, 47 SNP avec r2 > 0,7 et P ajusté > 0,01 ont été regroupés pour le test d’association.
En utilisant 5 000 permutations, un haplotype fortement lié avec une plage de 182 kb pourrait expliquer de manière significative la variation marron/jaune (χ2 = 37,7, P = 8,563e-06).
Le gène bien connu TYRP1 qui confère une couleur de plumage variable était situé à environ 531 kb de CCDC171.
La moyenne de la valeur LD entre eux a été estimée à <0,2.
Ces observations suggèrent que le gène contrôlant la couleur du plumage dans leur population était différent de TYRP1.
Les chercheurs ont ensuite choisi huit SNP significativement associés à la couleur du plumage, dont cinq sont situés dans le CCDC171, et ont conçu des amorces PCR pour amplifier ces marqueurs SNP afin de génotyper 100 cailles « marron » et 100 « jaunes » supplémentaires.
Fait intéressant, 99,75 % de ces SNP étaient cohérents entre le génotype et le phénotype, ce qui suggère que le gène CCDC171 contrôle la couleur du plumage des cailles.
Ils ont cloné le gène CCDC171 de la caille jaune et marron et ont découvert que ce gène codait différents transcrits chez la caille en fonction de la couleur du plumage.
Pour examiner la nature des variantes génétiques CCDC171, ils ont caractérisé les transcrits des allèles marron et jaune.
Le transcrit de l’allèle jaune était plus long que le transcrit marron (environ 232 pb) dans la région en amont du site d’initiation de la traduction du marron et présente une délétion (147 pb) à la position 787.
De plus, ils ont examiné l’expression différentielle de CCDC171. chez les cailles jaunes et marrons et ont trouvé qu’il n’y avait pas de différence significative entre les échantillons collectés (test t, P > 0,05).

Article rédigé et utilisant des sources traduites de l’Anglais par
Boris Nicoloff pour Bisou ma Caille© et Master Caille©
Droits de reproduction et de diffusion réservés © Bisou ma Caille© / Master Caille©
Sources et crédits des articles et photos utilisés dans cet article:
Population genomic data reveal genes related to important traits of quail
Yan Wu, Yaolei Zhang, Zhuocheng Hou, Guangyi Fan, Jinsong Pi, Shuai Sun, Jiang Chen, Huaqiao Liu, Xiao Du, Jie Shen, Gang Hu, Wenbin Chen, Ailuan Pan, Pingping Yin, Xiaoli Chen, Yuejin Pu, He Zhang, Zhenhua Liang, Jianbo Jian, Hao Zhang, Bin Wu, Jing Sun, Jianwei Chen, Hu Tao, Ting Yang, Hongwei Xiao, Huan Yang, Chuanwei Zheng, Mingzhou Bai, Xiaodong Fang, David W Burt, Wen Wang, Qingyi Li, Xun Xu, Chengfeng Li, Huanming Yang, Jian Wang, Ning Yang, Xin Liu, Jinping Du

